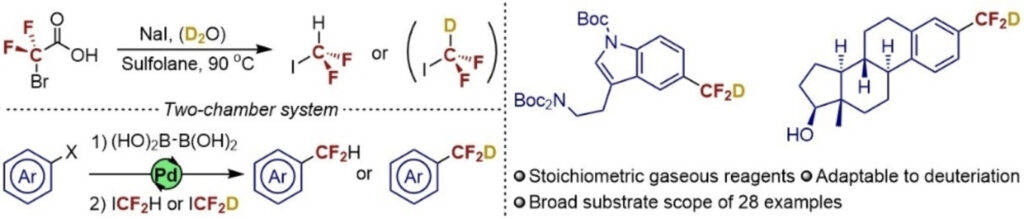
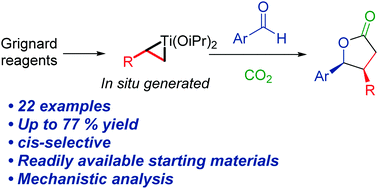
The research groups led by NordCO2 PIs Kim Daasbjerg and Troels Skrydstrup from Aarhus as well as Mårten Ahlquist from KTH Royal Institute of Technology have released a new paper where they explore the effect of additives for the electroreduction of CO2 to HCOOH, using a manganese electrocatalyst. Congratulations to all authors!

Title:
Promoting Selective Generation of Formic Acid from CO2 Using Mn(bpy)(CO)3Br as Electrocatalyst and Triethylamine/Isopropanol as Additives
Abstract:
Urgent solutions are needed to efficiently convert the greenhouse gas CO2 into higher-value products. In this work, fac-Mn(bpy)(CO)3Br (bpy = 2,2′-bipyridine) is employed as electrocatalyst in reductive CO2 conversion. It is shown that product selectivity can be shifted from CO toward HCOOH using appropriate additives, i.e., Et3N along with iPrOH. A crucial aspect of the strategy is to outrun the dimer-generating parent-child reaction involving fac-Mn(bpy)(CO)3Br and [Mn(bpy)(CO)3]− and instead produce the Mn hydride intermediate. Preferentially, this is done at the first reduction wave to enable formation of HCOOH at an overpotential as low as 260 mV and with faradaic efficiency of 59 ± 1%. The latter may be increased to 71 ± 3% at an overpotential of 560 mV, using 2 M concentrations of both Et3N and iPrOH. The nature of the amine additive is crucial for product selectivity, as the faradaic efficiency for HCOOH formation decreases to 13 ± 4% if Et3N is replaced with Et2NH. The origin of this difference lies in the ability of Et3N/iPrOH to establish an equilibrium solution of isopropyl carbonate and CO2, while with Et2NH/iPrOH, formation of the diethylcarbamic acid is favored. According to density-functional theory calculations, CO2 in the former case can take part favorably in the catalytic cycle, while this is less opportune in the latter case because of the CO2-to-carbamic acid conversion. This work presents a straightforward procedure for electrochemical reduction of CO2 to HCOOH by combining an easily synthesized manganese catalyst with commercially available additives.
Find a list of publications by NordCO2 members on our Publications page 🙂